Pharmaceutical regulation in the UK
A case study
31 October 2024
Reading time: 82 minutes

This work was commissioned as part of our report on lessons for AI regulation from the governance of other high-tech sectors. There are two other case studies:
Introduction
This paper considers five research questions. The research questions reflect active areas of discussion with regard to developing a regulatory framework for artificial intelligence. In applying these research questions to the established regulatory framework for medicines, we provide a basis for drawing lessons for designing AI regulation.
- What does pharmaceutical regulation seek to achieve?
- How does pharmaceutical regulation define and measure ‘public benefit’?
- What mechanisms does pharmaceutical regulation use to achieve its objectives?
- How are liability and compliance burdens distributed across the pharmaceutical value chain?
- How effective are these mechanisms at shaping industry behaviour?
This case study does not include regulation of biomedical research and largely excludes economic regulation (e.g. price control mechanisms, competition or procurement regulations).
Acronyms used in this paper
| ADR | Adverse drug reaction |
| CHM | Commission on Human Medicines |
| DWP | Department for Work and Pensions |
| DHSC | Department of Health and Social Care |
| EAMS | Early access to medicines scheme |
| eCTD | Electronic common technical document |
| EMA | European Medicines Agency |
| EU | European Union |
| ESMO | European Society of Medical Oncology |
| GMP | Good manufacturing practice |
| HMR | Human Medicines Regulations |
| ICER | Incremental cost effectiveness ratio |
| ILAP | Innovative Licensing and Access Pathway |
| LMIC | Low- and middle-income country |
| ESMO-MCBS | Magnitude of Clinical Benefit Scale |
| MHRA | Medicines and Healthcare products Regulatory Agency |
| MMDA | Medicines and Medical Devices Act |
| NICE | National Institute for Health and Care Excellence |
| NAS | New active substances |
| NHSE | NHS England |
| QALY | Quality-adjusted life years |
| RCT | Randomised controlled trials |
| RWE | Real-world evidence |
| R&D | Research and development |
| US FDA | U.S. Food and Drug Administration |
| WHO | World Health Organization |
| YCS | Yellow Card Scheme |
The context for life sciences regulation
The pharmaceutical industry is characterised by high research and development (R&D) investments, high potential risk to both drug developers and consumers, and the high potential social value of new and effective treatments. Pharmaceuticals are a highly regulated industry (though not uniquely high in regulations and comparable to other industries such as banking, tobacco, aircraft, utilities, communication and insurance).[1] Among new active substances (NAS), that is, medicines comprising entirely new active molecules, around 20–50 new medicines are approved by major high-income country regulators each year (Figure 1).[2] [3] Annual approval numbers for MHRA are not available, likely due to the recent separation from EMA processes.
Figure 1: Number of new active substances (NAS) approved by six regulatory authorities between 2011-2020 [4]
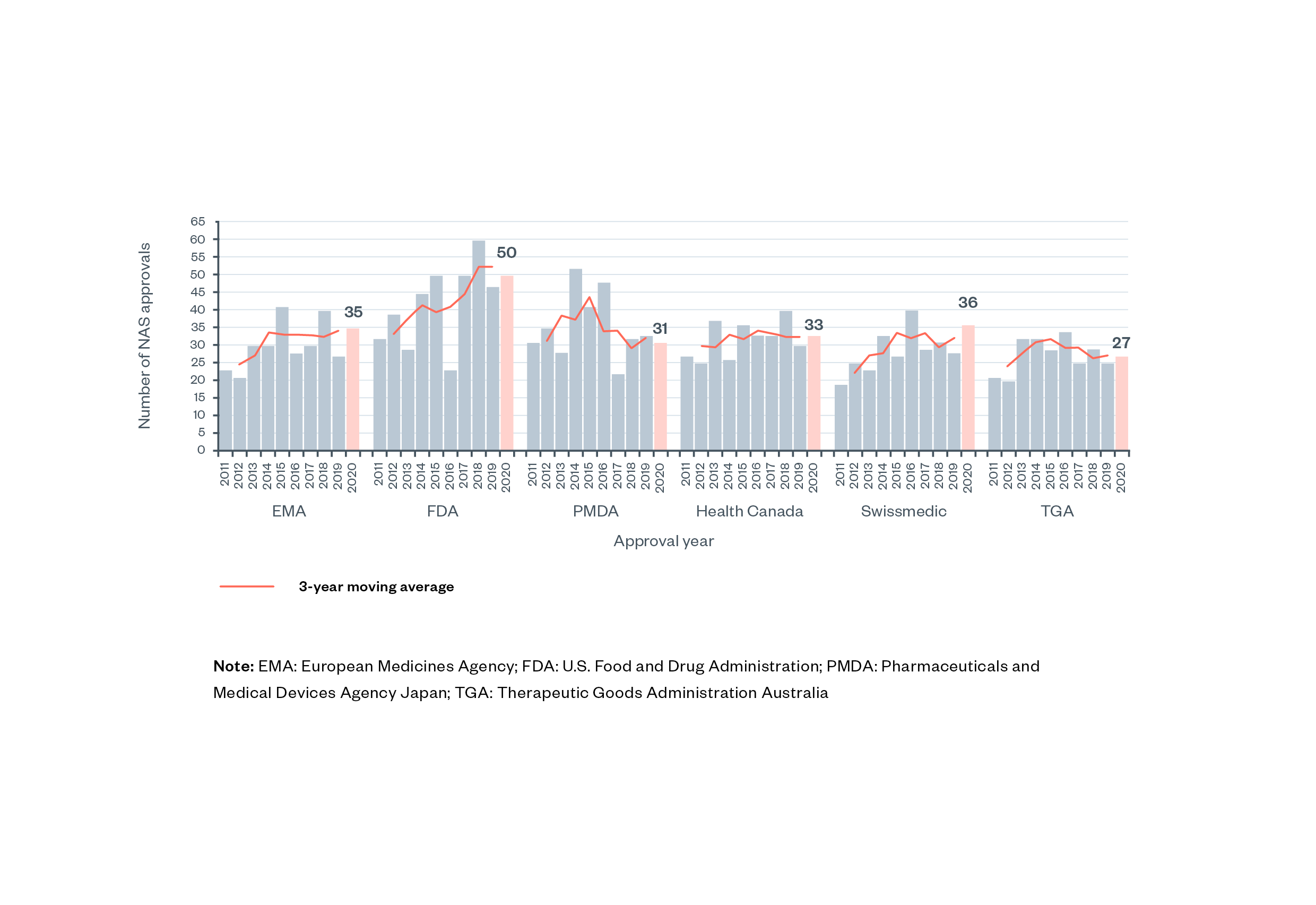
Note: EMA: European Medicines Agency; FDA: U.S. Food and Drug Administration; PMDA: Pharmaceuticals and Medical Devices Agency Japan; TGA: Therapeutic Goods Administration Australia.
Several groups of stakeholders are affected by UK pharmaceutical regulation, including:
- developers and manufacturers of medicines sold in the UK, including both UK-based pharmaceutical companies, which form a sector of domestic industry, as well as those of foreign pharmaceutical companies, whose products make up the majority of medicines used in the UK
- medicine distributors, who must, for example, ensure adequate storage conditions for medicines in transit
- people receiving medicines as part of medical treatment in the UK, whose health will suffer if regulation fails in its objectives and benefit if regulation is successful
- people working in the healthcare system, who have legal obligations placed on them by pharmaceutical regulation
- foreign governments that are influenced by the way the UK approaches pharmaceutical regulation, which, by extension, affects patients in foreign countries.
The UK domestic pharmaceutical industry
Pharmaceutical manufacturing represents a significant industrial sector in the UK. Gross value added (measured as economic output minus economic inputs, or revenue minus costs of production) from UK pharmaceutical manufacturing was £15 billion in 2020, making it the third largest sector of manufacturing in the UK, after ‘manufacture of food products, beverages and tobacco’ and ‘manufacture of basic metals and metal products’.[5] In terms of large, global multinational pharmaceutical companies, the UK is home to AstraZeneca and GlaxoSmithKline, which are the largest pharmaceutical companies in the UK by a wide margin.[6] In 2023, these two companies ranked ninth and tenth in market capitalisation, respectively, among pharmaceutical companies globally.[7] The UK consequently has an outsized impact on global pharmaceutical development. In 2015, it was reported that: ‘“approximately one-eighth of the world’s most popular prescription medicines [were] developed in UK laboratories.’[8] The UK pharmaceutical industry employs 73,000 people, of which around a third work in research and development (R&D).[9] [10]
As a buyer of medicines in the global market, the UK represents a relatively small market, comprising 2% of the global pharmaceutical market (the USA represents 43%, Europe 22%, China 8%, Japan 8% and India 1.5%).[11] [12] The UK imported £30.8 billion and exported £25.4 billion of pharmaceuticals in 2022, resulting in a negative trade balance. In a ranking of trade balances in pharmaceuticals by country, the UK fell from 4th place to 98th place globally over 2010–20.[13] Among generic medicines, which make up 75% of medicine consumption in the NHS, 80-90% are imported (no comparable figure is available for originator medicines – the first approved version of a medicine).[14] Generic medicines are clinically identical versions of originator medicines, generally sold by a competitor company.
Health R&D investments in the UK
The UK Government invests significant resources in health R&D, coming second to the USA among global comparator countries in terms of budget allocated to health R&D (UK budget allocations stood at 0.15% of GDP, compared with 0.23% of GDP in the USA, in 2020).[15]
Private-sector investments in pharmaceutical R&D, amounting to £5billion in 2020 (the most recent available data), make up one-fifth of R&D investments across all sectors in the UK.[16] As a point of comparison, in 2019 (the most recent available data), investments in medical and health R&D at UK higher education institutions amounted to £2.2 billion, private non-profit R&D was £899 million, and for UK government institutions was £267 million.[17]
Though this paper, due to its scope, focuses on the regulation of commercial pharmaceutical products, it is important to note that the UK public sector contributes significantly to global health R&D both through UK-based institutions and through overseas development aid.[18]
History of pharmaceutical regulation in the UK and relevant organisations
Laws regulating pharmaceuticals in England date back at least to 1540 and the Pharmacy Wares, Drugs and Stuffs Act, which empowered physicians to inspect apothecaries and destroy inadequate goods.[19]
The creation of pharmacopoeias – comprehensive lists of all medicines that can legally be created, sold, and used – were the next major step in regulation: the Pharmacopoeia Londinensis was first published in 1618, eventually becoming the British Pharmacopoeia in 1864.[20] [21] [22] Legislation in the seventeenth to nineteenth centuries did not include any mechanisms for testing whether substances were safe or effective, though they did introduce certification requirements for pharmacists, gave inspection powers to certain professional societies and limited the advertisements of ‘quack doctors’ and the adulteration of medicines.[23]
Historically, pharmaceutical regulation has often been reactive, triggered by events where products caused significant harms. The US Food, Drugs and Cosmetics Act (1938), which is generally seen as having provided the impetus for the development of the modern regulatory system in the USA, UK and Europe, was enacted in response to the scandal of sulphanilamide elixir poisoning in children.[24]
Two decades later, the thalidomide disaster, famously having prompted the creation of the US Food and Drug Administration, was also key in establishing modern pharmaceutical regulation in the UK.
Box 1: Thalidomide disaster
Thalidomide was a drug approved for sale for the treatment of morning sickness in pregnant people. Between 1956 and 1961, 12,000 children were born with limb defects and other congenital abnormalities, of which a third died in the first month of life. The manufacturer explicitly claimed that the medicine was safe for the foetus. At the time, there was ‘no legal obligation to demonstrate the safety or efficacy of their products before marketing them’.[25] [26]
The Medicines Act 1968 is the seminal piece of UK legislation establishing a central and far-reaching pharmaceutical regulatory system in the UK, coming into force in 1971.[27] Up until 1971, there was no centralised or comprehensive system for pharmaceutical regulation in the UK, no formal regulator and no body that could remove a drug from the market.[28] The USA had previously introduced, in 1938, safety requirements following the deaths of one hundred children after taking a syrup to treat strep throat.[29]
Pharmaceutical regulation in the UK largely took place through voluntary industry participation in schemes such as the Committee on Safety of Drugs (1963–71), which issued non-binding recommendations on the safety of medicines.[30] However, certain medicines and diseases were regulated by legislation created in the early twentieth century, including opiates, cocaine, vaccines, venereal diseases and cancer.[31] [32]
The Medicines Act 1968 established the ‘Licensing Authority’ and the Committee on Safety of Medicines (CSM). The Licensing Authority is a legislative concept, which was represented in practice by the Medicines Division of the Department of Health and Social Security (DHSS). CSM undertook review of product applications and made recommendations to DHSS regarding approval.
In 1989, the Medicines Division was ‘spun out’ of DHSS and renamed the Medicines Control Agency. In 2003, the Medicines Control Agency was merged with the Medical Devices Agency to become the Medicines and Healthcare products Regulatory Agency (MHRA).[33] [34]
The Medicines Act 1968 was also the first to introduce efficacy, safety and quality as criteria for approval. Before the Act came into force in 1971, pharmaceutical regulation was focused on quality (which essentially referred to chemical purity) of medicines, rather than safety and efficacy.[35] [36] The concept of ‘safety’ entails limiting the harms and risks of using a medicine to an acceptable level. The term ‘efficacy’ refers to the ability of a medicine to produce the desired beneficial effect. ‘Quality’ in this context refers to the physical purity of the product and correspondence to the parameters (e.g. ingredients) approved by the regulator.
These definitions are provided by the authors of this paper. We are not aware of an official definition of these terms in MHRA regulations or in UK legislation. A distinction is sometimes made between the words ‘efficacy’ and ‘effectiveness’, where the former refers to effects in a controlled experimental setting and the latter refers to effects in real-world use.
Current regulatory bodies
Figure 2: Relevant regulators for medicines and medical devices approval in England [37]

All medicines placed on the market in the UK require marketing authorisation, granted by the Medicines and Healthcare products Regulatory Agency (MHRA). Several independent advisory bodies support the work of MHRA, of which the Commission on Human Medicines (CHM) is most relevant for pharmaceuticals.
Applications for marketing authorisation are reviewed by the MHRA. In straightforward cases, generally concerning new generic products with equivalence to well-established products on the market or minor changes to the market authorisation for a previously authorised product, the MHRA can grant marketing authorisation without consulting the CHM. The MHRA will consult CHM on novel medicines (i.e., originator medicines or new active substances) and other complex applications. Additionally, all applications judged unsatisfactory by the MHRA are reviewed by CHM, which effectively acts as an ‘appeals’ process.[38]
Box 2: Functions of the Commission on Human Medicines, as set out in the Human Medicines Regulations 2012
- to advise the Health Ministers and the Licensing Authority on matters relating to human medicinal products including giving advice in relation to the safety, quality and efficacy of human medicinal products where either the Commission thinks it appropriate or where it is asked to do so
- to consider those applications that lead to Licensing Authority action as appropriate (i.e. where the Licensing Authority has a statutory duty to refer or chooses to do so)
- to consider representations made (either in writing or at a hearing) by an applicant or by a licence or marketing authorisation holder in certain circumstances
- to promote the collection and investigation of information relating to adverse reactions to human medicines for the purposes of enabling such advice to be given.[39]
Figure 3: Overview of the process for pharmaceutical approval in the UK
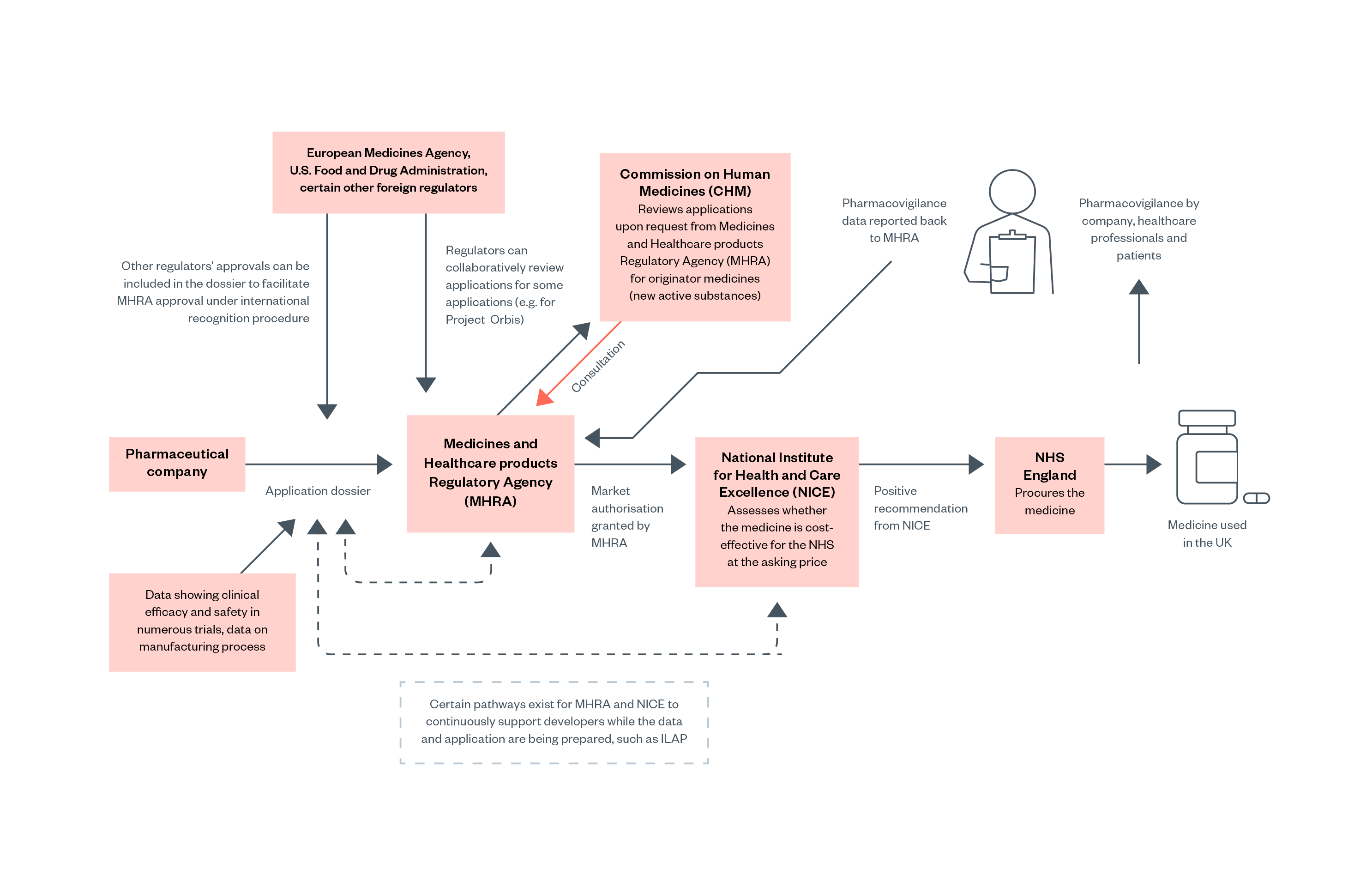
However, marketing authorisation by the MHRA does not necessarily mean that a medicine or product will be purchased or reimbursed by the NHS. The National Institute for Health and Care Excellence (NICE) then evaluates whether the medicine should be procured or reimbursed by NHS England (NHSE). NHSE is legally obligated to fund treatments recommended by NICE. Medicines can therefore have marketing authorisation and be available in the UK private market without being available on the NHS.[40]
Two key pieces of legislation relating to regulatory bodies following Brexit are the Human Medicines Regulations 2012 and the Medicines and Medical Devices Act 2021. These a ‘decouple’ the UK and EU regulatory systems. The Human Medicines Regulations 2012 incorporated much of EU law regarding medicines and incorporated many functions of the European Medicines Agency into the MHRA (these incorporated functions continue to be in force following Brexit, as ‘retained EU law’).[41] The Medicines and Medical Devices Act 2021provides mechanisms for the Department of Health and Social Care (DHSC) to change or introduce new regulations (which previously would have taken place mainly at the EU level).[42]
Defining the scope of what is a medicine
In UK and EU law, a product can fall into the definition of ‘medicinal products’ by two routes: either by being used for treatment or prevention, or by being presented as useful for treatment or prevention. For example, presenting a product in a form that appears to be a tablet or capsule can provide ‘strong evidence’ that falls under the definition of a ‘medicinal product’.[43] [44] [45]
Objectives of pharmaceutical regulation
The primary objectives of UK pharmaceutical regulation – namely the Human Medicines Regulations (2012) and Medicines and Medical Devices Act (2021)(Appendix) – are to ensure that the medicines used in the UK are effective, safe and of adequate manufacturing quality.
Secondary objectives include ensuring an enabling environment for the pharmaceutical industry, encouraging innovation, and positioning the UK as a global leader in health regulation.
Regulators must strike an appropriate balance between having a high degree of confidence in the safety of a product and enabling the product to enter the market as soon as is possible – while managing limited resources. To do this, regulators take a risk-stratification approach, meaning that certain types of products can be identified as needing greater scrutiny, while others can safely be reviewed at a lower level of scrutiny.
For example, generic medicines are approved with an shorter application dossier and are generally approved by the MHRA without consultation of its expert committees (see ‘Current regulatory bodies’ in the previous section). Originator medicines on the other hand, which represent entirely new compounds (molecules) that have not previously been approved, require a full application dossier and undergo detailed review by experts.
Regulators’ objectives in balancing benefits and harms
In one sense, pharmaceutical regulation relies on a binary system: a medicine either has marketing authorisation, meaning a government certification that it is safe and effective, or it does not.[46] In contrast, in clinical medicine, evaluations are rarely certain and are normally based on assessment of probabilities.[47]
As outlined above, regulators take a risk-stratification approach. The MHRA has an ‘explicit commitment to “risk-proportionate regulation”: this represents a significant intent to increase regulatory oversight on high-risk medical products, applications and indications [a medical condition that a medicine is used to treat], especially in relation to new technologies, with a parallel reduction in regulatory oversight on products where there is clear evidence of a lower risk to patients’. The MHRA does not define which products would be in this ‘high-risk’ group.[48]
Regulators are also, increasingly, introducing a spectrum of approval ‘states’. For example, under the Early Access to Medicines Scheme (EAMS; see Box 6 below), the MHRA may give a positive scientific ‘opinion’ on a medicine that has not yet been fully approved. Accelerated approval pathways (outlined in later sections) allow earlier approval of a medicine based on limited clinical data, with conditions attached to the approval, requiring drug companies to gather additional data.
The role of regulators in supporting R&D
Discussions around pharmaceutical regulation have long included concerns that regulation should consider the needs for enabling and increasing R&D. The 2023 ‘Pro-innovation Regulation of Technologies Review’ for life sciences, for example, gives recommendations on a ‘range of regulatory opportunities to enhance innovation in the health system’ and ‘explores issues which currently restrict the UK’s potential to be the best place in the world to develop, test, trial, manufacture and commercialise innovative new medical products’.[49] The MHRA describes its objective as ‘balancing’ its ‘responsibilities to maintain product safety and champion innovation’.[50]
One civil society interviewee for this paper argued that the focus of MHRA has increasingly moved to support the pharmaceutical industry’s needs, diluting the focus on patient safety. This is echoed in UK academics’ criticisms of new accelerated approval pathways.[51] In contrast, another interviewee from industry expressed concerns that MHRA was ‘restricted by its mandate to protect public health’.
The 2021 Medicines and Medical Devices Act (MMDA) makes explicit the regulatory objective of creating a favourable environment for industry by requiring DHSC to consider: ‘The likelihood of the relevant part of the United Kingdom being seen as a favourable place’ in which to undertake R&D, manufacture, or supply medicines when creating or changing medicines regulation (see Appendix). This also includes, implicitly, foreign companies or investors. The original draft wording of ‘the attractiveness of the relevant part of the United Kingdom as a place in which to conduct clinical trials or supply human medicines’ could be interpreted as a desire to compete internationally for the most business-favourable regulatory requirements.[52] [53] This provision was subject to controversy, with legal academics expressing concern that ‘placing the attractiveness of the UK in equal consideration to safety might lead to the latter being undermined’.[54] [55] The original wording was reworded after review in the House of Lords.
Interviewees from across academia, civil society and industry felt that international competition among regulators was accelerating in terms of implementing regulatory changes to increase the speed and ease of approval. Some expressed concerns of a potential ‘race to the bottom’ in safety standards. However, one interviewee from civil society argued that the policy subtext of international regulatory competition should not be taken in isolation, but instead contextualised within a wider effort by governments worldwide to be perceived as good places to do business. Regulation is a key domain but the language of competition is also observed in subsidies, taxation and intellectual property policy.
International relations
The MHRA’s goals are not only to define and administer regulations in the UK, but also to seek a leadership role within global regulation. This includes lobbying for other countries and regions to adopt regulation similar to the UK. For example, the MHRA’s 2018-2023 strategic vision included ‘[strengthening] our global positioning and reach[,] enhancing international partnerships, influencing emerging regulations and strengthening our commercial offering’.[56]
The All-Party Parliamentary Group on Global Health describes how the UK’s work in global health, including through the MHRA helps the UK ‘strengthen its influence and soft power’.[57]
Mechanisms of implementing pharmaceutical regulation
As discussed in preceding chapters, the approval of medicines requires proof of safety, efficacy and quality. Broadly, safety and efficacy are proven through clinical trial data, while quality is shown through quality standards, manufacturing process standards and quality control standards (also known as good manufacturing practice or ‘GMP’).[58]
Manufacturers demonstrate adherence to GMP (i.e. product quality) through, among other things, the submission of data from quality control testing and through in-person MHRA inspections of manufacturing sites.[59]
We do not go into details of pharmaceutical manufacturing regulation here. The focus is on regulation at the point of evaluating new active substances (NAS), which are more likely to provide useful analogies for AI regulation.
Data submission to the MHRA takes the form of an electronic common technical document (eCTD), a globally standardised format for clinical evidence submitted to regulators (with the MHRA directing applicants to European Medical Agency guidance on how to prepare the eCTD).[60] [61]
Current routes for MHRA approval of new active substances are outlined in Box 3 below. After approval, market authorisation must be renewed once after the first five years, after which authorisation lasts indefinitely. These first five years can therefore be considered a probationary period. However, the renewal process does not involve a detailed review of new evidence: if no serious concerns have been raised in the first five years, renewal is reportedly ‘essentially automatic’.[62] [63]
In 2021, as part of its Patient and Public Involvement Strategy, the MHRA announced its aim to ensure that two patient representatives are included in every decision-making committee.[64] [65] [66] While many interviewees expressed optimism about the spirit of more participatory and inclusive regulation, practical and structural concerns were also raised.
One interviewee expressed concern about the paucity of patient groups that are not reliant on funding from industry, and the potential for obscured conflicts of interest (COI).
Although industry involvement in patient groups requires COI disclosure, a civil society interviewee highlighted that patients representing a charity that is reliant on industry funding are generally not subject to the same COI standards – as they are not personally in receipt of funding. Another interviewee from a professional body expressed frustration in delays between the announcement of consultative process and the release of guiding frameworks.
Box 3: Current MHRA application routes for originator medicines (new active substances, NAS)
Multiple new MHRA application routes have been introduced recently, partly due to Brexit and partly due to continued efforts to streamline and accelerate review processes. For conciseness, we do not include procedures relating to Northern Ireland.
- National procedure
This is the default procedure for NAS or generics/biosimilars of already approved medicines. The procedure has a target of being completed in 150 days when submissions are of the correct format (a ‘high-quality submission’).[67] - Rolling review procedure
This is available for NAS only. This procedure allows applications to be made in separate ‘modules’ which can be submitted as they are ready, without waiting for a ‘full’ application dossier to be complete. This may create efficiencies on the side of the applicant, as parts of the application can enter regulatory review before all parts of the application dossier have been fully prepared.[68] - Innovative Licensing and Access Pathway (ILAP)
A pathway that developers can apply for while a drug is still in development. It is available for medicines that gain an ‘innovation passport’ designation. This is granted by the MHRA based on early data submitted by the developer. The criteria broadly covers medicines that are likely to present a therapeutic advance (as opposed to so-called ‘me too’ medicines, which have effects similarly to medicines already in use). MHRA and NICE provide support throughout the development process, with the aim of facilitating streamlined regulatory approval and overall accelerated market entry.[69] - International recognition procedure
Introduced on 1 January 2024, this is available for both NAS and generics/biosimilars and is based on a product having been approved by any ‘Reference Regulator’. These are authorities in Australia, Canada, Switzerland, Singapore, Japan, the USA or EU/EEA (both EMA and national authorities of the EU, Norway, Iceland, and Lichtenstein).[70] [71] - International work-sharing agreements
In both cases, one of the national application procedures listed above must still be followed.- Project Orbis
An international collaboration providing an approval pathway for oncology NAS or new indications – a medical condition that a medicine is used to treat. It allows joint concurrent or collaborative review by the US FDA and at least one other regulator from the UK, Australia, Canada, Singapore, Switzerland, Brazil or Israel. For MHRA review of an application as part of Orbis, the product must meet the criteria for ILAP – discussed above.[72] [73] - Access Consortium45
This procedure applies to NAS or generics/biosimilars for any indication. Two or more regulators from Australia, Canada, Singapore, Switzerland, and the UK can collaboratively process applications, while independently taking final regulatory decisions. The MHRA has been less involved in this collaboration compared to Project Orbis, above.[74] [75]
- Project Orbis
- Early access to medicines scheme (EAMS)
Though EAMS is not an approval pathway per se, it is included here as a related mechanism. Through the EAMS, the MHRA can issue scientific opinions on a medicine before it is approved. A positive opinion describes a favourable risk-benefit profile. The scientific opinion can support prescribers and patients in deciding to use an investigational medicine before approval. Acceptance for MHRA review through EAMS requires a ‘promising innovative medicine’ designation, granted by MHRA following an initial scientific meeting and review of early clinical data.[76] [77]
Regulatory reliance mechanisms
The MHRA’s international recognition procedure for approval (described in Box 3 above) has been described as ‘near automatic sign off’.[78] [79] [80] This is because medicines that have been approved by trusted overseas regulators (for example. the US FDA) can undergo a significantly shorter review process with MHRA.
The development of ‘recognition’ or ‘reliance’ procedures was historically focused on supporting access to medicines in low- and middle-income countries (LMICs). This included the World Health Organization (WHO) Prequalification Programme and the US FDA’s ‘tentative approval’ process. These procedures meant that international global health agencies and LMIC governments could rely on regulatory assessments in procuring medicines, when medicines had not been locally registered.[81] [82]
However, reliance or ‘recognition’ procedures are also often advocated as a mechanism for accelerating medicine approval by avoiding potentially duplicative review processes under different regulators. For example, early interim mechanisms created after Brexit enabled MHRA reliance on EMA approvals. In 2021, 68% of MHRA approvals for new medicines were done via EU reliance procedures.[83]
Reliance procedures also substantially reduce regulatory overhead for the industry. As regulators will often have different requirements regarding the format (e.g., language) of an application or the types of studies that must be performed, seeking approval in numerous countries significantly increases administrative burdens.
However, regulatory reliance procedures have been criticised as (too much) ‘outsourcing’ of decisions, which ‘could increase the number of low value medicines entering the UK market’.[84] ‘Outsourcing’ through regulatory reliance may diminish public-sector skills and capacity, with unintended consequences in other sectors. Regulatory reliance on another country may also mean ‘inheriting’ issues with that country’s approval processes. For example the US FDA’s accelerated approval pathway is based on limited clinical data and has been controversial. For example, due to using unvalidated ‘surrogate’ measures of effect and subsequent ‘confirmatory’ trials failing to show benefit.[85] Lastly, reliance procedures, by design, decrease the amount of expert scrutiny received by an application for a new medicine. Repeated assessments by different agencies may also offer confidence that nothing has been missed, by offering ‘another pair of eyes’.
Speed of approval
The regulatory process in England is relatively quick compared to other European countries. In terms of the time taken between market authorisation (MHRA approval) and availability to patients (availability through NHS England after a NICE recommendation), England came seventh out of thirty-seven countries in this metric in 2022.[86] Several caveats should be considered when interpreting such rankings. For example, in Germany, which tops the ranking, medicines are automatically approved for state reimbursement following market authorisation, pending an HTA assessment.
There are many studies comparing European Medical Agency (EMA) and FDA approval timelines, but limited data comparing the MHRA to the FDA. This is due to the relatively recent departure of the MHRA from the EMA centralised approval process.
Accelerated approval pathways, such as those offered by the US FDA, are now being mirrored in the UK through the new ILAP pathway (see Box 3 above and Figure 3). Concerns regarding such pathways are outlined below under the heading ‘Innovativeness’.
Pharmacovigilance
Pre-approval data, taking the form of randomised controlled trials , is generally considered more robust than post-approval data on adverse drug reactions (ADRs), which has mostly been based on voluntary reporting systems. It is believed that around 90% of ADRs go unreported.[87] It has been argued that ‘the decision to approve a medicine effectively signals an end to the gathering of meaningful and reliable information as to safety’.[88] Additionally, there have been cases where manufacturers have allegedly withheld concerning safety data from regulators, including for rofecoxib (an arthritis medicine) and paroxetine (an antidepressant).[89]
The MHRA is responsible for day-to-day monitoring of drug safety.[90] Pharmacovigilance in the UK takes place through two main routes: the Yellow Card Scheme (see Box 4 below), and pharmacovigilance databases maintained and monitored by the market authorisation holder.
As part of the electronic common technical document (eCTD) submitted for drug approval, pharmaceutical companies must include a ‘Pharmacovigilance System Summary’.[91] Drug companies are required to notify MHRA of any information that may be relevant to ‘the evaluation of the benefits and risks’ of a product as soon as practicable, including any (new) restrictions placed on a product in another country.[92] Drug companies ‘are obliged to notify [MHRA] of new information arising from any data source (except our database) which impact on the marketing authorisation’, and must have ‘signal detection systems [that] enable [them] to meet [their] requirements for cumulative signal detection across all available data sources’.[93] There are several known limitations of these schemes: reporting is only undertaken on patient, clinician or pharmacist initiative; proactive long-term follow-ups are not institutionally required; and some effects may take years to manifest and therefore are more challenging to link to drugs (e.g., teratogens).
Box 4: Pharmacovigilance schemes
Yellow Card Scheme (YCS)
The Yellow Card Scheme is an online database run by the MHRA, which relies on voluntary reports from healthcare professionals and the public.[94]
YCS data can be expected to be ‘noisy’, given that data is not collected in a systematic manner, is partly reported by non-professionals, and reported adverse events almost by definition have no proven causal link (they are suspected). In theory, the large volume of reports should allow for ‘signals’ to be detected where many reports of similar adverse events are made for a given product. This would prompt more rigorous investigation by the MHRA, specifically via the Commission on Human Medicines.[95] Due to the large volume of reports, MHRA uses data mining (statistical text analysis) to identify ‘signals’ of ‘disproportionality’ in reports submitted through the YCS, which are then further assessed by a group of experts.[96]
Black triangle scheme
The black triangle symbol indicates that the medicine is subject to ‘intense monitoring’, recognising that medicines are approved on the basis of clinical trials, which may not be representative of real-world use and patient experiences. It can be found next to the name of qualifying medicines in professional references (such as the British National Formulary) used by healthcare providers and in the patient information leaflet included in medication packaging.[97]
All new active substances are given a black triangle, as well as biological medicines, medicines given conditional approval and medicines for which the developer has been required to carry out additional studies. Black triangle status typically lasts 5 years.[98]
Conceptualising and measuring harms and benefits in life sciences regulation
In this chapter, we focus on how the benefits and harms of experimental treatments are assessed and quantified, particularly in the context of regulatory approval. We outline the role of randomised controlled trials, discuss the different measures (endpoints) that can be used, the emerging use of real-world evidence, and summarise the process for cost-effectiveness assessments of new pharmaceutical products in the UK.
Randomised controlled trials
High-quality randomised controlled trials (RCTs) are considered the gold-standard experimental design for assessing an experimental treatment.
The objective of the RCT design is to measure only the effect (and harms) of the experimental treatment, by minimising other factors (‘bias’) that would lead to different outcomes between the two groups. One key method for avoiding bias is randomisation. In the simplest design, trial participants (or subjects) are randomly divided into two or more groups, where one of the randomised groups receives the experimental treatment (the ‘intervention arm’), while the other group receives the established standard of care (the ‘control’ group or ‘placebo’ group).
Randomisation to different trial groups seeks to produce two maximally similar groups of patients while avoiding, for example, investigator bias in allocating patients perceived as more likely to have a good outcome to the experimental arm. Another approach is ‘blinding’. This is when steps are taken to ensure that neither the participants nor the investigators know which participants are receiving the treatment (for example, an experimental medicine versus a placebo).[99]
It should be noted that, in certain contexts, it may not be possible to undertake a trial that is randomised or blind. As one example, the very large ‘RECOVERY’ trial for COVID-19 treatments was conducted as ‘open-label’, i.e. not blind, meaning both the clinical teams and the patients knew which treatment was given. This was done due to the logistical difficulties of setting up placebos (for example, all patients would have had to receive an intravenous placebo, even if randomised to an oral therapy) and, in our view, likely also influenced by a desire to increase acceptability to patients in the pandemic context.[100]
Another example can be found in comparing a surgical procedure to a non-surgical intervention. In order to make the trial ‘blind’ to the patients, the triallists would need to perform ‘sham surgery’ on the placebo group, which would be considered unethical in many cases and is rarely done (though not entirely unprecedented).[101]
When RCTs are used for progressing through approval procedures, they are usually divided into three pre-approval phases and one post-approval phase (See Box 5 below). Medicines must be found to have satisfactory results in each phase in order to proceed to the next phase. Most medicines will undergo numerous trials at each phase. For example, multiple different Phase II trials may be undertaken for different indications, meaning, for example, for different diseases or different treatment combinations. ‘Success’ in each phase is determined by showing efficacy and safety that is at least equivalent (non-inferior), if not better (superior), to the existing standard treatment. The efficacy and safety are measured using various ‘endpoints’, outlined in the following section.
Box 5: Clinical trial phases[102] [103] [104]
Pre-clinical development (1–2 years): Identification of a broad pool of promising molecules, identification of the most promising among those, studies of the drug in vitro and in animals.
Phase I (~1 year): ‘First in human’ – a study in a small number of healthy volunteers to confirm safety.
Phase II (~2–3 years): A study in a moderate number of people with the relevant
disease/condition, to prove that the product has an effect and to identify a dose to use in final confirmatory trials.
Phase III (~2–3 years): A study in a large number of people with the relevant disease/condition, to compare the efficacy of the product to that of the standard of care.
Approval (~1 year): Approval is generally granted after at least one Phase III trial is completed, though some medicines are approved based on Phase II data alone.
Phase IV: Studies undertaken after approval, to investigate more nuanced questions regarding efficacy (such as optimum combinations with other medicines) and to further monitor safety.
In an analysis of 440 drugs approved in the USA between 2010 and 2020, the median time elapsed from Phase I through to gaining approval, was 8.3 years. But development times vary significantly between drugs: in the aforementioned analysis, the range was 2.7 to 43.7 years.[105]
Clinical endpoints and clinical effectiveness
A range of clinical measures or targeted outcomes, known as ‘endpoints’, can be used to measure the efficacy and safety of medicines. One example is mortality: what is the rate of survival (over a set period of time) in one group of patients compared to another group? This is known as a ‘hard’ endpoint, i.e., one that has a clear binary state. Other endpoints can include biochemical markers, such as those measured in blood tests; imaging findings, such as the change in the size of tumours as measured in scans; or quality-of-life measures, such as self-reported symptom severity.
The choice of endpoints in clinical trial has been a key area of evolution for evidence-based medicine and the biomedical industry over recent decades. The appropriateness of endpoints used in trials has frequently been the focus of debate. For example, where endpoints are considered ‘proxy’ or ‘surrogate’ endpoints, rather than ‘hard’ measures of outcomes, regulators and other stakeholders may consider evidence for a medicine’s effectiveness less strong.[106]
Often, ‘proxy’ endpoints, which are generally easier and/or faster to measure, are used. This includes, as one example, using blood cholesterol levels as a proxy measure of cardiovascular risk: while high blood cholesterol in itself does not cause any symptoms, the developers may argue that, due to the established link between cholesterol levels and death from cardiovascular causes, a new medicine that lowers blood cholesterol should reduce the risk of death.
It has been shown that, although regulatory approval has often been based on ‘surrogate’ endpoints, surrogate endpoints often do not correlate to clinically important effects such as survival time.[107] [108] The implication of these findings is that, as a result of methodological changes aimed at making drug development and approval faster and less costly, some medicines that are approved are not as effective and/or safe as clinicians and patients might expect.
Due to the importance of the choice of endpoints for a trial, regulators have published guidance on recommended endpoints. The European Medicines Agency, for example, provides detailed guidance on the investigational plan, including recommended endpoints, by disease.[109]
In modern RCTs, a large number of endpoints are collected, many of which represent continuous, rather than binary, variables. A continuous variable is a variable that can take any numerical value (e.g. blood sugar level). A binary variable is a variable that can only take two values (e.g. living or dead).
This raises the question of what a clinically significant magnitude of effect is. Statistical analysis may also reveal statistically significant differences between the experimental and ‘control’ groups that may not be clinically significant. For example, if radiological imaging reveals a statically significant difference in tumour size after treatment with a new medicine, but this does not translate to longer survival or symptom change, the mere radiological finding could be considered to lack clinical meaning. Grading systems have been designed to help aggregate data from different endpoints into scores of clinical benefit. One example is the Magnitude of Clinical Benefit Scale (ESMO-MCBS) developed by the European Society of Medical Oncology (ESMO).[110]
Patient-reported outcomes are a category of clinical endpoints that generally represent treatment effects (or harms) that cannot be objectively measured. This includes, for example, symptom burden, other quality-of-life metrics, and satisfaction with treatment.[111] Patient-reported outcomes have increasingly been used in trials, and several regulators offer guidance and standards on the use of such endpoints in evidence supporting applications.[112]
Real-world evidence
The pharmaceutical industry and regulators have been developing ways to use ‘real-world evidence’ (RWE) to partially replace traditional RCTs. RWE represents data collected outside of RCTs, such as data from patient registries (large databases following patient outcomes during the course of ‘normal’ treatment), insurance databases, patient networks, and wearables.[113]
RWE can be used to support the approval of new indications (treatment uses) for medicines that have already been approved; to optimise treatment combinations and dosing regimens; and to study effects in certain sub-populations. It is argued that RWE can be used to represent or complement the ‘control’ group in a RCT, allowing RCTs to be smaller.[114]
Regulators have approved medicines using experimental designs that use RWE to represent a control arm, though regulatory developments are ongoing.[115] In studies of medicines for rare diseases, the ‘control’ arm can be based on RWE because of ethical concerns of randomising some patients to a ‘control’ arm—for example, where this would mean not giving any active treatment (as no approved treatment exists).[116] [117]
Economic benefits and economic harms
The National Institute of Health and Care Excellence (NICE) makes judgements on the cost-effectiveness of medicines compared to available alternatives, with NHS England legally required to procure medicines recommended by NICE. NICE therefore plays a key role in allocative distributions within the NHS and has a significant effect on the UK pharmaceutical market.[118] NICE ‘balance[s] the need to achieve the most overall benefit for the greatest number of people, with the need to ensure fairness and respect for individual choice’, based on ‘social value judgements’.[119]
Box 6: Incremental cost effectiveness ratio (ICER)

For NICE, cost is expressed in GBP and effect is expressed in quality-adjusted life years (QALY), meaning the ratio is expressed in GBP per QALY gained. A quality-adjusted life year is a measure of a technology’s effect on a patient’s combined quantity and quality of life, where 1 QALY represents one additional year of life in perfect health and 0 QALYs represents death.[120]
NICE uses a guiding threshold of 20-30,000 GBP/QALY, above which technologies are less likely to be recommended.[121] While NICE reports that, since 2000, 84% of its appraisals have been ‘positive’, a significant proportion of ‘positive’ appraisals recommended use of the medicine for a narrower group of patients than was indicated in the original market authorisation.[122]
Two interviewees, from academia and civil society, expressed concern of ‘constant pressure’ for NICE to raise the cost-effectiveness threshold, and that NICE was insufficiently politically insulated to withstand such pressure. Another issue, raised by an academic interviewee, was increased confidentiality demands made by industry around pricing, which reduces the usefulness of NICE evaluations as a mechanism for public transparency around the costs and benefits of medical technologies.
Innovativeness
The level of efficacy that is sufficient to achieve market authorisation may simply be superiority to a placebo in a selected group of patients or non-inferiority to the existing, established treatment (standard of care).[123]
This allows registration of ‘me too’ medicines – medicines that add to the list of available options but do not represent a therapeutic advance – as there is no requirement for the medicine to be superior to the standard of care.
Only a small proportion of new medicines that are approved every year offer significant clinical advances. The majority of new approvals are medicines that are similar to the existing standard of care, often including closely related drugs within the same class of compounds. A range of different assessment systems (e.g. therapeutic value rating systems used by the Canadian, French, and German governments) show that less than half or as little as 3% of new medicine approvals represent significant added therapeutic value, depending on the time period and grading system.[124] [125] [126] [127] [128] [129]
Additionally, evidence from the USA suggests that programmes designed to accelerate regulatory review processes for medicines have not been effective in identifying medicines with significant added therapeutic benefits: drugs undergoing expedited review in two-thirds of cases do not perform better than available alternatives and have a higher rate of safety events and withdrawals from market.[130] [131] [132] [133] [134] (See also ‘Speed of approval’ in the preceding chapter.)
Burden of liability and compliance along the value chain
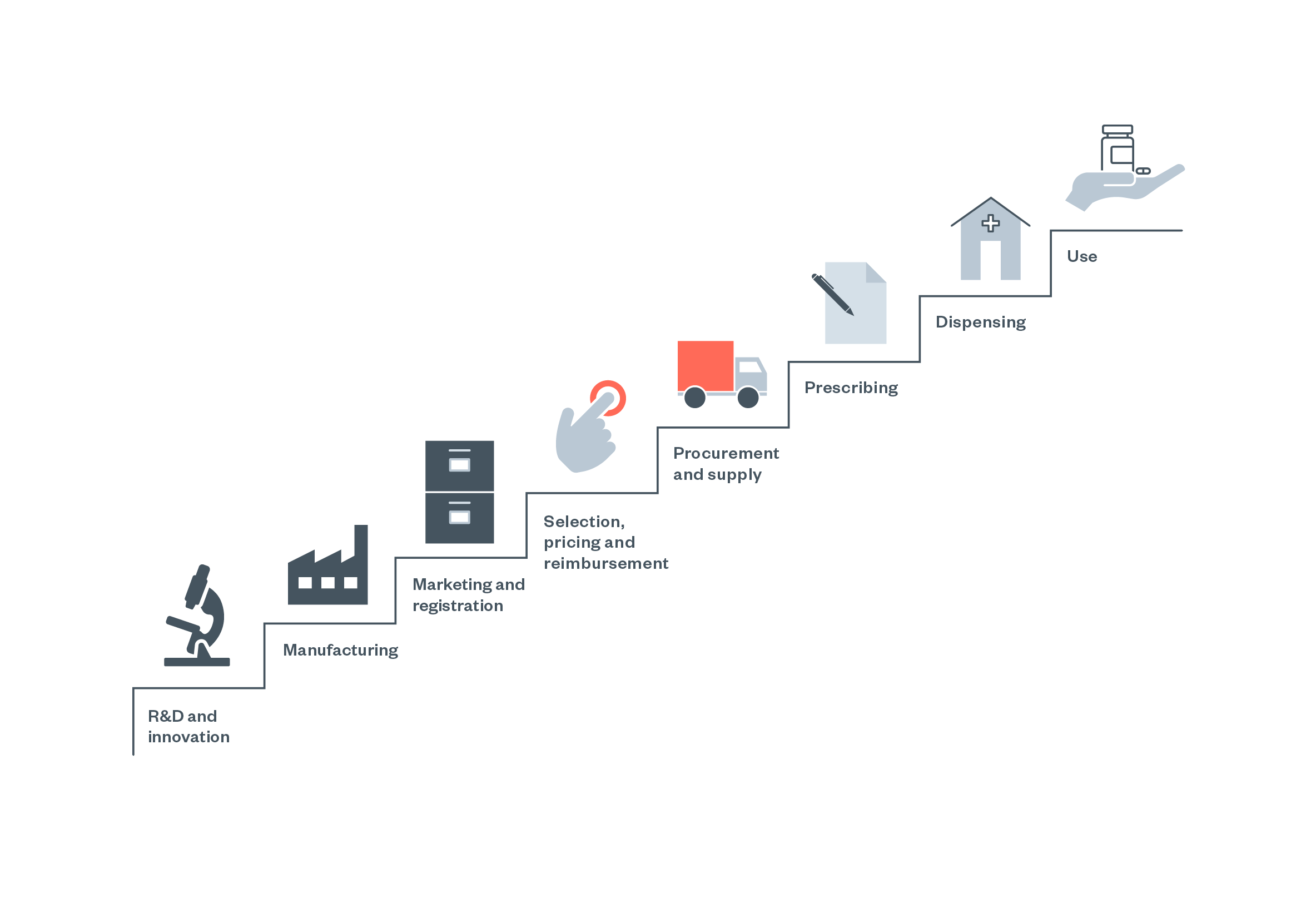
This chapter provides an overview of pharmaceutical liability along the value chain (Figure 3) under different areas of law in the UK. Overall, it is argued that chances of an injured person (i.e. a patient taking a medicine) successfully bringing a claim against a drug manufacturer are ‘slim, at best, and non-existent at worst’ in the UK. This is in contrast with higher success rates in, for example, the USA, as also highlighted by one academic with a legal background.[135] The barriers for potential claimants in the UK are outlined in the following subsections, and mainly concern challenges in using tort law or the Consumer Protection Act.
Different regulatory jurisdictions implement different liability regimes: contract law, tort law and/or no-fault compensation mechanisms. Liability regimes generally apply to all products, with specific liability regimes for pharmaceuticals being rare globally and absent in the UK.[136]
Figure 4: The pharmaceutical ‘value chain’ as conceptualised by the WHO [137]
Tort law (negligence)
Under established UK tort law, manufacturers have a duty of care towards their consumer to take reasonable care to ensure their products are safe.[138] [139] Doctors and pharmacists similarly have a duty under the tort of negligence.[140] Difficulties may arise in proving that the harm done was due to negligent manufacture or prescribing rather than the underlying illness (with the example of the selective serotonin reuptake inhibitor (SSRI) antidepressant class being associated with an increased risk of suicide, as also is the depressive disorder being treated), or due to a particular drug if the patient is using numerous medicines.[141]
Patients are, in theory, warned in numerous ways of the risk of adverse drug reactions (ADRs). Doctors should advise patients of potential ADRs (though the list of possible ADRs is generally very large and established practice is to highlight only common ADRs or rare-but-serious ADRs). Leaflets that are required to be supplied with medicines will also list ADRs. This may also make it difficult for a patient to show that they were not adequately warned of risks.[142]
The 2015 Montgomery v Lanarkshire case overturned the preceding Bolam test (Bolam v Friern Hospital Management Committee, 1957) by increasing the duty of risk disclosure from Bolam’s standard of negligence defined as not being ‘in accordance with a practice accepted as proper by a responsible body of medical men skilled in that particular art’ to a higher standard of a ‘duty to take reasonable care to ensure that the patient is aware of any material risks involved in any recommended treatment, and of any reasonable alternative or variant treatments’.[143] [144] One interviewee from a legal background argued that the Montgomery judgement would make negligence claims through doctors or prescribers more attractive than claims against manufacturers.
Breach of statutory duty
A person suing a regulator for harm from a drug that the regulator had approved would have to prove a ‘relationship of proximity’ between them and the regulator and that it would be fair, just and reasonable for the regulator to have a duty of care to the individual patient.[145] [146] Furthermore, a claimant would have to prove that it was unreasonable to grant marketing authorisation for the medicine. As the MHRA grants authorisations (for new medicines) based on the recommendations of a group of experts, this would be difficult.[147]
The Consumer Protection Act and strict product liability
The Consumer Protection Act 1987 established ‘strict liability’, meaning that the producer of a product is liable for any defects in the product, with ‘defect’ defined as occurring ‘[i]f the safety of the product is not such as persons generally are entitled to expect’. It remains in force post-Brexit, though it will likely be more difficult to enforce a claim against a non-UK-based trader.[148]
Professor Emily Jackson (London School of Economics) has argued that: ‘The Consumer Protection Act, despite its name, has proved to be a remarkably consumer-unfriendly piece of legislation, and there have as yet been no successful cases against pharmaceutical companies’.[149]
Jackson continues that ‘few successful cases [were] brought under this piece of legislation and hardly any cases involving medicines’ because the definition of a ‘defect’ is problematic in the context of medicines, where adverse reactions will be expected to occur in a proportion of patients for almost any medicine.[150] Successful consumer protection actions for medicines have been far more common in the USA.[151]
One legal academic interviewee noted heterogeneity across the United Kingdom in settlement practices, observing that companies were more likely to settle in Scotland than England because i) Scottish courts had historically been more sympathetic to claimants, and ii) Scotland has a small population compared to England, so the cost of settling compared favourably to the cost of legal fees.
No-fault compensation (redress) schemes
There has been increasing interest in establishing no-fault compensation schemes in Europe and the United States.[152]
In no-fault compensation schemes, the claimant is required only to show harm from the product, without needing to prove the manufacturer’s fault.[153] [154] These schemes also implement an administrative process in determining compensation rather than using adversarial litigation.[155] [156] They are ‘typically presented as compromises designed to achieve reasonable compensation for the greater number of victims, rather than full compensation for very few’.[157] [158] [159]
Examples of such schemes in the UK include the Thalidomide Trust, considered a ‘ground-breaking event in British legal history, involving settlement of claims in circumstances in which legal liability was unclear but social responsibility was evident’.[160] There are also schemes for those affected by the ‘infected blood scandal’ in which people receiving transfusions for haemophilia were infected with hepatitis C virus and/or HIV, and a fund for people who contracted variant Creutzfeldt-Jakob disease (‘mad cow disease’ in humans) from contaminated government-manufactured human growth hormone.[161] [162] Vaccine damage schemes exist, but are beyond the scope of this paper.[163]
Box 7: Examples of compensation schemes
Thalidomide Trust
The lawsuit against the UK distributor of thalidomide was settled privately in 1968, with the High Court approving the agreement on liability and the form of the settlement. The distributor agreed to pay 40% of the amount claimed in the suit, plus inflation, with amounts scaled to the degree of disability. The original settlement covered 65 individuals. Further claims were brought for hundreds of other individuals. After years of negotiation, a new settlement was reached in 1973, which provided individual settlements for the claimants, as well as a charitable trust for the payment of damages to all children accepted as having injuries due to thalidomide (based on a set of eligibility criteria). The Thalidomide Children’s Trust was established under court order, funded with a £14 million donation from the distributor. The initial plan was for payments to be made out of the interest on this investment, amended in the mid-1990s to allow the distribution of the capital. Payments into the trust, made by the distributor (and later companies that acquired the distributor) have continued at least until 2022, with an available amount for distribution calculated yearly. The UK government has also made contributions to the Trust. Distribution is based on eligibility criteria and the degree of disability, as assessed by the Trust.[164]
HIV/HCV infected blood scandal
Compensation is provided by two limited companies and three charities each with partially overlapping eligibility criteria for claims. No negligence of fault has been legally established, and ‘the establishment of the scheme ended attempts to sue the government’.[165] All five entities are fully financed by the UK Government.[166]
A 2015 All-Party Parliamentary Group explained the background for this arrangement:
‘Rather than administering the support for those affected directly through the Department of Health or Department for Work and Pensions (DWP), successive governments – in part to avoid the implication of legal responsibility for infecting them – have opted to deliver it through external entities set up by the Department of Health solely for the purpose of supporting them. These trusts are funded solely by ‘ex gratia’ payments from both the Department of Health and, in certain instances, the devolved governments. Those affected, and their families, have to register with the trusts and prove their eligibility in order to receive payments from them.’[167]
Contract law
As patients do not buy drugs directly from the manufacturer, the chance of successfully suing under contract law in the UK is ‘extremely remote’[168] and also rare in the EU.[169]
Doctors or pharmacists could be sued for harm (under contract law) if they directly sell a medicine of inadequate quality to a patient, or if the patient is harmed through a contra-indication of which the seller was aware.[170] However, this is a rare scenario in the UK, where direct sales are uncommon. Direct sales would apply to medicines bought in the private sector and over-the-counter medicines.[171] Some EU countries extend contract protection to third parties (i.e., those that are not parties to the original contract) or to all intermediate sellers along the sales chain.[172]
Regulatory compliance defence
In a ‘regulatory compliance’ defence under the Consumer Protection Act, a manufacturer can argue that they are not liable for damages caused by a defective medicine if they have properly complied with the regulatory regime,[173] where the defect was ‘due to compliance’ with laws.[174]
State of the art defence
In a ‘state of the art’ defence under the Consumer Protection Act, a manufacturer may argue that they are not liable for damages if the ‘state of scientific and technical knowledge at the time was not such that a producer […] might be expected to have discovered the defect’.[175] It has been argued that, because much of ADR detection occurs (and is expected to occur) after regulatory approval of the product, this shifts the risk of injury from the producer onto the injured person.[176]
Effectiveness of the regulatory environment in shaping business practices
In many senses, UK pharmaceutical regulation is powerful in shaping business practices, as businesses must comply with a range of regulatory requirements in order to receive permission to place a product on the market. On the other hand, regulation is ‘co-produced’ by the industry and the UK government. For example, Wiktorowicz et al. describe the UK model of pharmaceutical regulation as a ‘bargaining’ model based in corporatism, wherein the ‘regulator negotiates with industry to achieve mutual adjustment’.[177]
This is contrasted with, for example, a ‘pluralist’ and ‘managerial discretion’ model in the USA, which provides more opportunities for ‘external challenges’ to the regulatory regime, for example, through litigation, and where the regulator is (more) guided by its own mandate and analyses.[178] One illustration of the ‘bargaining’ versus ‘pluralist/managerial discretion’ distinction is in the approach to post-market data gathering (pharmacovigilance). In the UK’s ‘bargaining’ model, pharmacovigilance is largely the responsibility of the company, while in the USA’s ‘managerial discretion’ model, the FDA is more active in investigating safety issues independently.[179]
Davis and Abraham have linked much of the UK’s current model of regulatory engagement with pharmaceutical industry to neoliberal reforms starting with the Thatcher administration.[180] The newly formed Medicines Control Agency increased consultations with industry and included their interests in its mission statement, while negotiating licensing fees with the industry that made the Agency ‘one of the richest regulatory agencies in Europe’.[181] The MCA’s model was soon imitated by other European countries, such as Germany and Sweden.[182]
Despite potential benefits from technical discussions between industry and regulators, there is often conflict between the interests of pharmaceutical companies and the principles of approving only safe and effective medicines.[183] [184] [185] [186] In response to recent government announcements regarding a ‘pro-innovation’ regulatory environment, leading UK academics have expressed concerns that: ‘[T]he MHRA fails to recognise the potential for conflict between its role as a public health agency and its promise to “reduce regulatory burden” to support “today’s brilliant life sciences industry” and “boost growth”’.[187]
Effects of regulatory requirements on competition in pharmaceuticals
Pharmaceutical development requires significant early investments and involves significant investment risks. The overall costs of developing one medicine are generally considered to be in the range US$1-2 billion, factoring in the costs of failed R&D projects and the cost of capital.[188] These costs are primarily driven by the high costs and time required for running multiple clinical trials and the relatively high risk of failure at each phase of development. Additionally, drug development requires complex infrastructure such as laboratories, manufacturing facilities and clinical facilities. Some of these functions can often be outsourced: for example, clinical research organisations (CROs) can run clinical trials.
The need for high up-front, at-risk investments and the need for significant infrastructure and know-how means there is a high barrier for new drug developers wanting to enter the sector. As a result, small developers (biotechs) will commonly sell their drug candidate molecules to large pharmaceutical companies (or the entire company may be acquired) early in the development pathway: often at a point where only animal and in vitro studies have been completed.
Post-marketing requirements
The effectiveness of regulation in shaping business practices is less clear with regard to post-marketing requirements, compared to the requirements for initial approval. A significant proportion of medicines are approved ‘conditionally’, meaning that companies are required to undertake additional studies on certain aspects of the medicine (generally, safety) as a condition of approval.
While data on UK-specific compliance with such requirements are not available, a study of European Medicines Agency (EMA) cancer medicine approvals found that 47% of post-marketing requirements were not completed on time.[189] These reflections are especially relevant in the context of ‘accelerated’ approval pathways, in which approvals are often ‘conditional’ as described above. One recent (2024) example of the MHRA withdrawing conditional marketing authorisation is the drug crizanlizumab (sold as ‘Adakveo’), for which Phase III study data did not confirm clinical benefit for sickle cell disease.[190]
Industry fees as funding for regulators
The MHRA has undergone reorganisation, significant workforce cuts, and cost-cutting in recent years.[191] From its creation in 2003 and 2022, the MHRA was a ‘government trading fund’, meaning an agency that is self-funding.[192] [193] Predecessors to the MHRA had also been majority-funded by industry fees, with changes instituted by Prime Minister Margaret Thatcher requiring all costs to be covered by fees.[194] Reportedly, the change in status in 2022 to allow greater contributions from the Treasury was related to reductions in the MHRA’s revenue from fees following Brexit.
This change means that ‘MHRA is not able to retain and rely on cash reserves to manage areas of under-recovery as it has done previously. This means that any over or underspend will directly impact the financial position of the Department of Health and Social Care, HM Treasury, and have implications for other Government Departments.’[195]
One interviewee with regulatory expertise highlighted that the physical proximity of the EMA (which was based in London before Brexit) to the MHRA enabled easy consultations between professionals in the two agencies and the loss of this has been felt at the MHRA after the EMA moved to Amsterdam.
In 2021, 86% of MHRA funding came from industry fees, compared to 89% for the EMA, 96% for the Australian regulator TGA and 65% for the US FDA.[196] This reliance on industry funding has attracted a range of concerns in many countries. Scientific experts serving on regulators’ advisory committees also often receive industry funding. A general industry tax or levy (i.e., payments not linked to individual applications) has been proposed as an alternative method of funding regulators.[197] Relatedly, an interviewee from industry also noted that a ‘revolving door’ between regulators and industry is well-recognised,[198] [199] and conflicts of interest for experts working with regulators are ‘tamed’ or ‘managed’ rather than being prohibited.[200]
Pressures to shorten regulatory review times
There are clear benefits to accelerating regulatory review, both for patients, who may receive an effective treatment sooner, and to the industry, whose revenues on a given medicine will begin sooner and last longer. Requests for regulatory processes to be sped up are a mainstay of industry commentary.
The MHRA aims to ‘compete’ with regulators in other countries with regard to speed of approval, aiming to achieve ‘competitive timescales for the UK determination of every type of application in comparison with other international regulators’.[201] Lexchin argues that this reflects ‘regulatory commercialism as many drug regulatory agencies now compete with each other for business and are subject to the “tests of the market” [within] a broader transformation of the state in the era of capitalist globalisation and the emergence of “competition states” which prioritise meeting the demands of global economic competitiveness’.[202]
Relatedly, it has been argued that shortening target approval timelines is a type of deregulation.[203] It has been argued that the UK’s relatively small share of the global pharmaceutical market (2.4%) may mean that pharmaceutical companies can afford to forgo or delay launching medicines in the UK if it is disincentivised by regulatory factors.[204] These concerns reflect the ‘tests of the market’ mentioned above.
‘Locking in’ of interim measures
In ‘early-access’ schemes, which are proliferating globally, the use of an experimental medicine in the health system is permitted before ‘full’ approval by the relevant regulator. In the UK, the main example is the Early Access to Medicines Scheme (EAMS), outlined above in Box 6, which allows access to experimental medicines with a ‘promising investigative medicine’ (PIM) designation many months before ‘full’ approval.[205]
In discussions with interviewees, it was argued that there may be ‘locking in’ or ‘ratcheting up’ effects: once an early-access mechanism becomes widely used, it is difficult for a regulatory system to stop the use of such a mechanism becoming the norm. Once a medicine has entered clinical use, it is often difficult to withdraw it (even if later clinical data suggest it is not effective), for example, where patients have already been established on a long-term treatment plan. Policymakers may also find it difficult to argue that medicines shouldn’t ‘be made available sooner’, due, among other things, to pro-innovation bias.[206] It was also argued in our interviews, by a health economics expert, that the increasing discussions of using real-world evidence as part of medicines approvals may allow an overly permissive regulatory standard to become established:
‘[W]hat really worries me is the notion of real-world data and collecting evidence being used as an excuse to approve things that aren’t cost-effective – it’s a fig leaf. If you didn’t have the political will [at the time of review] to reject it or demand evidence, or demand a reduction in price[,] you are not going to have political will when that product is [already] diffused widely.
Appendix: Objectives of UK regulation
Medicines and Healthcare products Regulatory Agency (MHRA)
MHRA’s definition of the agency’s core purpose to use ‘scientific expertise, support for innovation and the risk-proportionate regulation of medical products, to protect and improve public health across the UK.[207]
MHRA’s responsibilities as expressed on its website
‘Our responsibilities are to:
- ensure medicines, medical devices and blood components for transfusion meet applicable standards of safety, quality and efficacy (effectiveness)
- secure safe supply chain for medicines, medical devices and blood components
- promote international standardisation and harmonisation to assure the effectiveness and safety of biological medicines
- educate the public and healthcare professionals about the risks and benefits of medicines, medical devices and blood components, leading to safer and more effective use
- enable innovation and research and development that is beneficial to public health
- collaborate with partners in the UK and internationally to support our mission to enable the earliest access to safe medicines and medical devices and to protect public health’[208]
Human Medicines Regulations (2012)
Criteria for market authorisation:
‘The licensing authority may grant the application only if, having considered the application and the accompanying material, the authority thinks that—
(a) the applicant has established the therapeutic efficacy of the product to which the application relates;
(b) the positive therapeutic effects of the product outweigh the risks to the health of patients or of the public associated with the product;
(c) the application and the accompanying material complies with regulations 49 to 55; and
(d) the product’s qualitative and quantitative composition is as described in the application and the accompanying material.’[209]
Medicines and Medical Devices Act (2021)
Elements DHSC must consider when creating or changing medicines regulation:
- the safety of human medicines;
- the availability of human medicines;
- the likelihood of the relevant part of the United Kingdom being seen as a favourable place in which to—
- carry out research relating to human medicines,
- conduct clinical trials, or
- manufacture or supply human medicines.[210]
Priorities for UK regulatory science in health (2021 survey of 145 stakeholders)
- Flexibility: the capability of regulations to adapt to novel products and target patient outcomes.
- Co-development: collaboration across sectors, e.g. patients, manufacturers, regulators, and educators working together to develop appropriate training for novel product deployment.
- Responsiveness: the preparation of frameworks which enable timely innovation required by emerging events.
- Speed: the rate at which new products can reach the market.
- Reimbursement: developing effective tools to track and evaluate outcomes for ‘pay for performance’ products education and professional development.[211]
Footnotes
[1] O Al-Ubaydli and PA McLaughlin, ‘A Numerical Database on Industry-Specific Regulations for All US Industries and Federal Regulations, 1997–2012’ (Mercatus Center, November 2014 <www.mercatus.org/research/working-papers/regdata>.
[2] Centre for Innovation in Regulatory Science, ‘New Drug Approvals in Six Major Authorities 2011–2020: Focus on Facilitated Regulatory Pathways and Worksharing’ (R&D Briefing 81, June 2021) <www.cirsci.org/wp-content/uploads/dlm_uploads/2021/06/CIRS-RD-Briefing-81-6-agencies-v5.pdf>.
[3] MP Hofer and others, ‘Regulatory Policy and Pharmaceutical Innovation in the United Kingdom after Brexit: Initial Insights’ (2022) 9, 1011082 Frontiers in Medicine.
[4] ibid.
[5] Office for National Statistics, ‘The Industrial Analyses (Data Tables)’ (31 October 2022) <www.ons.gov.uk/economy/grossdomesticproductgdp/compendium/unitedkingdomnationalaccountsthebluebook/2022/theindustrialanalyses> accessed 28 January 2024.
[6] Statista, ‘Largest Pharmaceutical Companies by Market Capitalization in the United Kingdom as of June 6, 2023’ <www.statista.com/statistics/1345251/uk-largest-pharmaceutical-companies-by-market-capitalization> accessed 26 January 2024.
[7] Drug Discovery & Development, ‘2023 Pharma 50: The 50 Largest Pharma Companies in the World’ <www.drugdiscoverytrends.com/2023-pharma-50-largest-companies> accessed 27 January 2024.
[8] All-Party Parliamentary Group on Global Health, ‘The UK’s Contribution to Health Globally: Benefiting the Country and the World’ (29 June 2015) <www.thet.org/wp-content/uploads/2017/08/The-UKs-contribution-to-health-globally_FULL-1.pdf>.
[9] ABPI, ‘Why Work in the Industry?’ <www.abpi.org.uk/careers/why-work-in-the-industry> accessed 20 March 2024.
[10] ABPI, ‘Biopharma Employment Figures in UK’ <www.abpi.org.uk/facts-figures-and-industry-data/uk-pharmaceutical-market/biopharma-employment-figures-in-uk> accessed 20 March 2024.
[11] ABPI, ‘Shares of the Global Pharmaceutical Markets’ <www.abpi.org.uk/facts-figures-and-industry-data/global-pharmaceutical-market/shares-of-the-global-pharmaceutical-markets> accessed 20 March 2024.
[12] N Fleming, ‘Critics Wary over Plans to Fast-Track UK Drug-Approval Model’ (2023) Nature (27 July 2023) <www.nature.com/articles/d41586-023-02426-7>.
[13] ABPI, ‘International Trade’ <www.abpi.org.uk/international-trade-and-ip/international-trade> accessed 20 March 2024.
[14] House of Commons Business, Energy and Industrial Strategy Committee, ‘The Impact of Brexit on the Pharmaceutical Sector: Ninth Report of Session 2017–19’ (8 May 2018) <https://publications.parliament.uk/pa/cm201719/cmselect/cmbeis/382/382.pdf>.
[15] Office for Life Sciences, ‘Life Sciences Competitiveness Indicators 2023’ (13 July 2023) <www.gov.uk/government/publications/life-sciences-sector-data-2023/life-sciences-competitiveness-indicators-2023> accessed 29 December 2023.
[16] Office for Life Sciences, ‘Life Sciences Competitiveness Indicators 2023’ (n 15).
[17] Ibid.
[18] All-Party Parliamentary Group on Global Health, ‘The UK’s Contribution to Health Globally’ (n 8).
[19] RE Ferner and JK Aronson, ‘Medicines Legislation and Regulation in the United Kingdom 1500–2020’ (2023) 89, 80 British Journal of Clinical Pharmacology.
[20] Ibid.
[21] M Wood, ‘A “Weapon Dressed as a Book”: The Pharmacopoeia Londinensis’ (Royal College of Physicians, 8 June 2018) <https://history.rcplondon.ac.uk/blog/weapon-dressed-book-pharmacopoeia-londinensis> accessed 18 March 2024.
[22] E Jackson, Law and the Regulation of Medicines (Oxford: Hart Publishing, 2012).
[23] Ferner and Aronson, ‘Medicines Legislation’ (n 19).
[24] PR Lee and J Herzstein, ‘International Drug Regulation’ (1986) 7, 217 Annual Review of Public Health.
[25] Jackson, Law and the Regulation of Medicines (n 22).
[26] H Teff, ‘Regulation under the Medicines Act 1968: A Continuing Prescription for Health’ (1984) 47, 303.
[27] Jackson, Law and the Regulation of Medicines (n 22).
[28] Independent Medicines and Medical Devices Safety Review, ‘Annex H: History of Regulation Supporting Information (2020) <https://immdsreview.org.uk/downloads/Annexes/Annex-H-History-of-Regulation.pdf>.
[29] Jackson, Law and the Regulation of Medicines (n 22).
[30] Independent Medicines and Medical Devices Safety Review, ‘Annex H’ (n 28).
[31] Ferner and Aronson, ‘Medicines Legislation’ (n 19).
[32] Independent Medicines and Medical Devices Safety Review, ‘Annex H’ (n 28).
[33] Ibid.
[34] Ferner and Aronson, ‘Medicines Legislation’ (n 19).
[35] Ibid.
[36] Jackson, Law and the Regulation of Medicines (n 22).
[37] ‘Pro-Innovation Regulation of Technologies Review: Life Sciences and the Government Response’ (GOV.UK) <https://www.gov.uk/government/publications/pro-innovation-regulation-of-technologies-review-life-sciences> accessed 10 October 2024
[38] Department of Health, ‘Triennial Review of the Commission on Human Medicines: Review Report’ (26 March 2015) <https://assets.publishing.service.gov.uk/media/5a80c9bced915d74e6230715/chm-review-report.pdf>.
[39] Lee and Herzstein, ‘International Drug Regulation’ (n 24).
[40] Jackson, Law and the Regulation of Medicines (n 22).
[41] DHSC, ‘Explanatory Memorandum to the Human Medicines (Amendment etc.) (EU Exit) Regulations 2019’ <www.legislation.gov.uk/ukdsi/2019/9780111179185/pdfs/ukdsiem_9780111179185_en.pdf>.
[42] Medicines and Medical Devices Act 2021 <www.legislation.gov.uk/ukpga/2021/3>.
[43] Jackson, Law and the Regulation of Medicines (n 22).
[44] ‘Criminal Proceedings against Leendert van Bennekom. Judgement of the Court (Fifth Chamber of 30 November 1983’ (EUR-Lex, document 61982CJ0227) <https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A61982CJ0227> accessed 22 December 2023.
[45] The Human Medicines Regulations 2012. <www.legislation.gov.uk/uksi/2012/1916>.
[46] Jackson, Law and the Regulation of Medicines (n 22).
[47] A Cahan and others, ‘Probabilistic Reasoning and Clinical Decision-Making: Do Doctors Overestimate Diagnostic Probabilities?’ (2003) 96, 763 QJM: An International Journal of Medicine.
[48] MHRA, ‘MHRA Corporate Plan 2023–26’ <https://assets.publishing.service.gov.uk/media/64a41dd54dd8b3000f7fa452/230628_MHRA_Corporate_Plan_A4_CC_V14.pdf> accessed 23 December 2023.
[49] HM Government, ‘Pro-innovation Regulation of Technologies Review: Life Sciences’ (May 2023) <https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1159408/Life_sciences_report_-_Pro-innovation_Regulation_of_Technologies.pdf>.
[50] MHRA, ‘MHRA Corporate Plan 2023–26’ (n 48).
[51] H Naci, R Forrest and C Davis, ‘Putting Patients First in Medicines Regulation?’ (2021) 375, n2883 BMJ.
[52] L Downey and others, ‘Medicines and Medical Devices Act 2021 and Uncertain Regulatory Futures’ (University of Birmingham) <www.birmingham.ac.uk/research/perspective/medical-devices-act-regulation.aspx> accessed 9 December 2023.
[53] UK Parliament, ‘Medicines and Medical Devices Bill (HC Bill 90), as Introduced’ (13 February 2020) <https://publications.parliament.uk/pa/bills/cbill/58-01/0090/cbill_2019-20210090_en_2.htm#pt1-l1g1> accessed 9 December 2023.
[54] Ibid.
[55] Downey and others, ‘Medicines and Medical Devices Act’ (n 52).
[56] MHRA, ‘MHRA Corporate Plan 2023–26’ (n 48).
[57] All-Party Parliamentary Group on Global Health, ‘The UK’s Contribution to Health Globally’ (n 8).
[58] Jackson, Law and the Regulation of Medicines (n 22).
[59] MHRA, ‘Good Manufacturing Practice and Good Distribution Practice’ (27 December 2020) <www.gov.uk/guidance/good-manufacturing-practice-and-good-distribution-practice#overview> accessed 4 February 2024.
[60] MHRA, ‘Apply for a Licence to Market a Medicine in the UK’ (17 January 2024) <www.gov.uk/guidance/apply-for-a-licence-to-market-a-medicine-in-the-uk> accessed 29 January 2024.
[61] ICH, ‘ICH Electronic Common Technical Document: eCTD v3.2.2 Specification and Related Files’ <www.ich.org/page/ich-electronic-common-technical-document-ectd-v322-specification-and-related-files>.
[62] Jackson, Law and the Regulation of Medicines (n 22).
[63] House of Commons Health Committee, ‘The Influence of the Pharmaceutical Industry: Fourth Report of Session 2004–05’ (22 March 2005) <https://publications.parliament.uk/pa/cm200405/cmselect/cmhealth/42/42.pdf>.
[64] Naci, Forrest and Davis, ‘Putting Patients First’ (n 51).
[65] DHSC, ‘Government Response to the Report of the Independent Medicines and Medical Devices Safety Review’ (26 July 2021) <https://assets.publishing.service.gov.uk/media/60fea6688fa8f50432ab9217/IMMDS_Review_-_Government_response_-_220721.pdf>.
[66] MHRA, ‘Proposed Patient and Public Involvement Strategy 2020–25’ (24 May 2021) <www.gov.uk/government/consultations/mhra-patient-involvement-strategy-consultation/proposed-patient-and-public-involvement-strategy-2020-25> accessed 28 December 2023.
[67] MHRA (Medicines and Healthcare Products Regulatory Agency), ‘150-Day Assessment for National Applications for Medicines’ (5 January 2024) <www.gov.uk/guidance/guidance-on-150-day-assessment-for-national-applications-for-medicines> accessed 29 January 2024.
[68] MHRA, ‘Rolling Review for Marketing Authorisation Applications’ (31 December 2020) <www.gov.uk/guidance/rolling-review-for-marketing-authorisation-applications> accessed 29 January 2024.
[69] MHRA, ‘Innovative Licensing and Access Pathway’ (27 January 2023) <www.gov.uk/guidance/innovative-licensing-and-access-pathway> accessed 29 January 2024.
[70] MHRA, ‘International Recognition Procedure’ (5 January 2024) <www.gov.uk/government/publications/international-recognition-procedure/international-recognition-procedure> accessed 29 January 2024.
[71] MHRA, ‘International Recognition Procedure: Supplementary Information’ (5 January 2024) <www.gov.uk/government/publications/international-recognition-procedure/international-recognition-procedure-supplementary-information> accessed 29 January 2024.
[72] MHRA, ‘Project Orbis’ <www.gov.uk/guidance/guidance-on-project-orbis> accessed 29 January 2024.
[73] MP Lythgoe and others, ‘From the European Medicines Agency to Project Orbis: New Activities and Challenges to Facilitate UK Oncology Drug Approval following brexit’ (2023) 24, e150 The Lancet Oncology.
[74] MHRA, ‘Access Consortium’ (6 November 2023) <www.gov.uk/guidance/access-consortium> accessed 29 January 2024.
[75] Lythgoe and others, ‘From the European Medicines Agency to Project Orbis’ (n 73).
[76] MHRA, ‘Apply for the Early Access to Medicines Scheme (EAMS)’ (18 April 2023) <www.gov.uk/guidance/apply-for-the-early-access-to-medicines-scheme-eams> accessed 29 January 2024.
[77] DJ O’Connor, K McDonald and SP Lam, ‘Earlier Patient Access: The UK Early Access to Medicines Scheme (EAMS)’ (2017) 1 Medicine Access @ Point of Care.
[78] MHRA, ‘International Recognition Procedure’ (n 70).
[79] MHRA, ‘International Recognition Procedure: Supplementary Information’ (n 71).
[80] M Limb, ‘UK to Give “Near Automatic Sign Off” for Treatments Approved by “Trusted” Regulators’ (2023) 380, p633 BMJ.
[81] A Vaz and others, ‘WHO Collaborative Registration Procedure using Stringent Regulatory Authorities’ Medicine Evaluation: Reliance in Action?’ (2022) 15, 11 Expert Review of Clinical Pharmacology.
[82] EFM ’t Hoen and others, ‘A Quiet Revolution in Global Public Health: The World Health Organization’s Prequalification of Medicines Programme’ (2014) 35, 137 Journal of Public Health Policy.
[83] Hofer and others, ‘Regulatory Policy’ (n 3).
[84] MP Lythgoe and R Sullivan, ‘Outsourcing UK Regulatory Decisions: A Double-Edged Sword?’ (2023) 402, 24 The Lancet.
[85] Ibid.
[86] M Newton and others, ‘EFPIA Patients W.A.I.T. Indicator 2022 Survey’ (IQVIA, April 2023) <www.efpia.eu/media/s4qf1eqo/efpia_patient_wait_indicator_final_report.pdf>.
[87] N Raj and others, ‘Postmarket Surveillance: A Review on Key Aspects and Measures on the Effective Functioning in the Context of the United Kingdom and Canada’ (2019), 10 Therapeutic Advances in Drug Safety
[88] Jackson, Law and the Regulation of Medicines (n 22).
[89] Ibid.
[90] Department of Health, ‘Triennial Review’ (n 38).
[91] MHRA, ‘Marketing Authorisation Pre-Submission Checklist’ <https://assets.publishing.service.gov.uk/media/5a7eefdfed915d74e33f35c5/Pre-submission_checklist.pdf> accessed 29 January 2024.
[92] MHRA, ‘Guidance on Pharmacovigilance Procedures’ (28 October 2022) <www.gov.uk/government/publications/guidance-on-pharmacovigilance-procedures/guidance-on-pharmacovigilance-procedures#signal-detection> accessed 3 February 2024.
[93] Ibid.
[94] MHRA, ‘Yellow Card: Information’ <https://yellowcard.mhra.gov.uk/information> accessed 4 February 2024.
[95] Department of Health, ‘Triennial Review’ (n 38).
[96] MHRA Yellow Card Scheme, ‘Guidance on Adverse Drug Reactions’ <https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/949130/Guidance_on_adverse_drug_reactions.pdf> accessed 4 February 2024.
[97] MHRA, ‘Black Triangle Scheme: New Medicines and Vaccines Subject to EU-Wide Additional Monitoring’ <https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/396808/Black_Triangle_Scheme_-_new_medicines_and_vaccines_subject_to_EU-wide_additional_monitoring.pdf> accessed 3 February 2024.
[98] Ibid.
[99] EC Zabor, AM Kaizer and BP Hobbs, ‘Randomized Controlled Trials’ (2020) 158, S79 Chest.
[100] L Peto, P Horby and M Landray, ‘Establishing COVID-19 Trials at Scale and Pace: Experience from the RECOVERY Trial’ (2022) 86, 100901 Advances in Biological Regulation.
[101] BR Wolf and JA Buckwalter, ‘Randomized Surgical Trials and “Sham”’ Surgery: Relevance to Modern Orthopaedics and Minimally Invasive Surgery’ (2006) 26, 107 Iowa Orthopedic Journal.
[102] National Institute for Health and Care Research, ‘Clinical Trials Guide’ <www.nihr.ac.uk/documents/clinical-trials-guide/20595> accessed 7 January 2024.
[103] N Parvizi and K Woods, ‘Regulation of Medicines and Medical Devices: Contrasts and Similarities’ (2014) 14, 6 Clinical Medicine (London).
[104] National Research Council (US) Panel on Handling Missing Data in Clinical Trials, ‘Appendix A. Clinical Trials: Overview and Terminology’ in The Prevention and Treatment of Missing Data in Clinical Trials (Washington, DC: National Academies Press, 2010) <www.ncbi.nlm.nih.gov/books/NBK209903> accessed 7 January 2024.
[105] DG Brown and others, ‘Clinical Development Times for Innovative Drugs’ (2021) 21, 793 Nature Reviews Drug Discovery.
[106] C McLeod and others, ‘Choosing Primary Endpoints for Clinical Trials of Health Care Interventions’ (2019) 16, 100486 Contemporary Clinical Trials Communications.
[107] D Dawoud and others, ‘Raising the Bar for Using Surrogate Endpoints in Drug Regulation and Health Technology Assessment’ (2021) 374, n2191 BMJ.
[108] A Walia, A Haslam and V Prasad, ‘FDA Validation of Surrogate Endpoints in Oncology: 2005–2022’ (2022) 34, 100364 Journal of Cancer Policy.
[109] European Medicines Agency, ‘Clinical Efficacy and Safety Guidelines’ <www.ema.europa.eu/en/human-regulatory-overview/research-and-development/scientific-guidelines/clinical-efficacy-and-safety-guidelines> accessed 31 January 2024.
[110] NI Cherny and others, ‘ESMO Magnitude of Clinical Benefit Scale version 1.1’ (2017) 28, 2340 Annals of Oncology.
[111] NL Crossnohere and others, ‘International Guidance on the Selection of Patient-Reported Outcome Measures in Clinical Trials: A Review’ (2021) 30, 21 Quality of Life Research.
[112] Ibid.
[113] AE Baumfeld and others, ‘Trial Designs using Real-World Data: The Changing Landscape of the Regulatory Approval Process’ (2020) 29, 1201 Pharmacoepidemiology and Drug Safety.
[114] Ibid.
[115] Ibid.
[116] DC Klonoff, ‘The New FDA Real-World Evidence Program to Support Development of Drugs and Biologics’ (2020) 14, 345 Journal of Diabetes Science and Technology.
[117] N Mahendraratnam and others, ‘Understanding Use of Real-World Data and Real-World Evidence to Support Regulatory Decisions on Medical Product Effectiveness’ (2022) 111, 150 Clinical Pharmacology & Therapeutics.
[118] V Charlton, ‘NICE and Fair? Health Technology Assessment Policy under the UK’s National Institute for Health and Care Excellence, 1999–2018’ (2020) 28, 193 Health Care Analysis.
[119] National Institute for Health and Care Excellence, ‘Our Principles’ <www.nice.org.uk/about/who-we-are/our-principles> accessed 11 December 2023.
[120] Charlton, ‘NICE and Fair?’ (n 118).
[121] Ibid.
[122] National Institute for Health and Care Excellence, ‘Technology Appraisal Data: Appraisal Recommendations’ <www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-technology-appraisal-guidance/data/appraisal-recommendations> accessed 4 February 2024.
[123] Jackson, Law and the Regulation of Medicines (n 22).
[124] B Wieseler, N McGauran and T Kaiser, ‘New Drugs: Where Did We Go Wrong and What Can We Do Better?’ (2019) 366, I4340 BMJ.
[125] VN Vokinger and others, ‘Therapeutic Value of Drugs Granted Accelerated Approval or Conditional Marketing Authorization in the US and Europe From 2007 to 2021’ (2022) 3, e222685 JAMA Health Forum.
[126] VN Vokinger and others, ‘Therapeutic Value of First versus Supplemental Indications of Drugs in US and Europe (2011–20): Retrospective Cohort Study’ (2023) 382, e074166 BMJ.
[127] Prescrire, ‘Prescrire’s Ratings of New Drugs in 2022: A Brief Review’ (2023) 43, 146 Revue Prescrire.
[128] L Osipenko and others, ‘The Origin of First-in-Class Drugs: Innovation versus Clinical Benefit’ (2024) 115, 342 Clinical Pharmacology & Therapeutics.
[129] VN Vokinger and others, ‘Therapeutic Value Assessments of Novel Medicines in the US and Europe, 2018–2019’ (2022) 5, e226479 JAMA Network.
[130] Naci, Forrest and Davis, ‘Putting Patients First’ (n 51).
[131] TJ Hwang and others, ‘Association between FDA and EMA Expedited Approval Programs and Therapeutic Value of New Medicines: Retrospective Cohort Study’ (2020) 7(371), m3434 BMJ.
[132] SR Mostaghim, JJ Gagne and AS Kesselheim, ‘Safety Related Label Changes for New Drugs after Approval in the US through Expedited Regulatory Pathways: Retrospective Cohort Study’ (2017) 358, j3837 BMJ.
[133] NS Downing and others, ‘Postmarket Safety Events among Novel Therapeutics Approved by the US Food and Drug Administration between 2001 and 2010’ (2017) 317, 1854 JAMA.
[134] C Frank and others, ‘Era of Faster FDA Drug Approval Has also Seen Increased Black-Box Warnings and Market Withdrawals’ (2014) 33, 1453 Health Affairs.
[135] Jackson, Law and the Regulation of Medicines (n 22).
[136] M Martín-Casals, ‘Some Comparative Remarks on Pharmaceutical Product Liability’ (2018) 20, 5 PPL.
[137] ‘WTO | Promoting Access to Medical Technologies and Innovation: Intersections between Public Health, Intellectual Property and Trade (Second Edition)’ <https://www.wto.org/english/res_e/publications_e/who-wipo-wto_2020_e.htm> accessed 10 October 2024
[138] ‘M’Alister (or Donoghue) (Pauper) Appellant; and Stevenson Respondent’ (1932) UKHL 100 <www.bailii.org/uk/cases/UKHL/1932/100.html> accessed 22 December 2023.
[139] Jackson, Law and the Regulation of Medicines (n 22).
[140] Ibid.
[141] Ibid.
[142] Ibid.
[143] ‘Montgomery v Lanarkshire Health Board’ (2015) <www.supremecourt.uk/cases/docs/uksc-2013-0136-judgment.pdf> accessed 21 March 2024.
[144] ‘Bolam v Friern Hospital Management Committee’ 1957 <www.supremecourt.uk/cases/docs/uksc-2013-0136-judgment.pdf>.
[145] B Dimond, ‘Legal Regulation Mechanisms in the Control of Medicines’ (2014) 12(9), 560 British Journal of Nursing.
[146] Jackson, Law and the Regulation of Medicines (n 22).
[147] Ibid.
[148] L Conway, ‘Brexit: UK Consumer Protection Law’ (House of Commons Briefing Paper, 21 May 2021 <https://researchbriefings.files.parliament.uk/documents/CBP-9126/CBP-9126.pdf>.
[149] Jackson, Law and the Regulation of Medicines (n 22).
[150] Ibid.
[151] Ibid.
[152] S Macleod and others, Redress Schemes for Personal Injuries (Oxford: Hart Publishing, 2017).
[153] Martín-Casals, ‘Some Comparative Remarks’ (n 136).
[154] Macleod and others, Redress Schemes (n 152).
[155] Martín-Casals, ‘Some Comparative Remarks’ (n 136).
[156] Macleod and others, Redress Schemes (n 152).
[157] Martín-Casals, ‘Some Comparative Remarks’ (n 136).
[158] Macleod and others, Redress Schemes (n 152).
[159] D Jutras, ‘Alternative Compensation Schemes from a Comparative Perspective’ in M Bussani and AK Sebok (eds), Comparative Tort Law (Cheltenham: Edward Elgar Publishing, 2015).
[160] Macleod and others, Redress Schemes (n 152).
[161] Ibid.
[162] C Dyer, ‘Families Win Right to Compensation over CJD’ (1998) 316, 1625 BMJ.
[163] Macleod and others, Redress Schemes (n 152).
[164] Ibid.
[165] Ibid.
[166] All-Party Parliamentary Group on Haemophilia and Contaminated Blood, ‘Inquiry into the Current Support for Those Affected by the Contaminated Blood Scandal in the UK’ (January 2015) <https://haemophilia.org.uk/wp-content/uploads/2017/02/appg_hcb_fr.pdf.pdf>.
[167] Ibid.
[168] Jackson, Law and the Regulation of Medicines (n 22).
[169] Martín-Casals, ‘Some Comparative Remarks’ (n 136).
[170] Jackson, Law and the Regulation of Medicines (n 22).
[171] Ibid.
[172] Martín-Casals, ‘Some Comparative Remarks’ (n 136).
[173] Jackson, Law and the Regulation of Medicines (n 22).
[174] Arnold & Porter, ‘Global Practice Guide: Life Sciences’ (Chambers, April 2019) <www.arnoldporter.com/-/media/files/perspectives/publications/2019/04/chambers-life-sciences-2019-uk.pdf?rev=9583b5b6ec2548e8850a971c2f9fc051&sc_lang=en&hash=A0749A624913E337F2447D52646181F6>.
[175] Jackson, Law and the Regulation of Medicines (n 22).
[176] Martín-Casals, ‘Some Comparative Remarks’ (n 136).
[177] M Wiktorowicz, J Lexchin and K Moscou, ‘Pharmacovigilance in Europe and North America: Divergent Approaches’ (2012) 75, 165 Social Science & Medicine.
[178] Ibid.
[179] Ibid.
[180] C Davis and J Abraham, Unhealthy Pharmaceutical Regulation: Innovation, Politics and Promissory Science (Houndmills, Basingstoke, and New York: Palgrave Macmillan, 2013).
[181] Ibid.
[182] Ibid.
[183] Jackson, Law and the Regulation of Medicines (n 22).
[184] Naci, Forrest and Davis, ‘Putting Patients First’ (n 51).
[185] House of Commons Health Committee, ‘The Influence of the Pharmaceutical Industry’ (n 63).
[186] Davis and Abraham, Unhealthy Pharmaceutical Regulation (n 180).
[187] Naci, Forrest and Davis, ‘Putting Patients First’ (n 51).
[188] Knowledge Portal on Innovation and Access to Medicines, ‘Costs of Pharmaceutical R&D’ <www.knowledgeportalia.org/costs-r-d> accessed 20 March 2024.
[189] A Cherla and others, ‘Post-Marketing Requirements for Cancer Drugs Approved by the European Medicines Agency, 2004–2014’ (2022) 112, 846 Clinical Pharmacology & Therapeutics.
[190] MHRA, ‘Class 2 Medicines Recall: Novartis Pharmaceuticals UK Limited, Adakveo 10 mg/ml Concentrate for Solution for Infusion, EL(24)A/06’ (21 February 2024) <www.gov.uk/drug-device-alerts/class-2-medicines-recall-novartis-pharmaceuticals-uk-limited-adakveo-10-mg-slash-ml-concentrate-for-solution-for-infusion-el-24-a-slash-06> accessed 8 August 2024.
[191] G Iacobucci, ‘UK Drug Regulator Formalises Plan to Cut Staff in Response to Brexit Income Loss’ (2021) 375, n3058 BMJ.
[192] DHSC and MHRA, ‘Framework Agreement between the DHSC and the Medicines and Healthcare Products Regulatory Agency’ (15 March 2016) <www.gov.uk/government/publications/dh-and-mhra-framework-agreement/framework-agreement-between-dhsc-and-the-medicines-and-healthcare-products-regulatory-agency>.
[193] MHRA, ‘MHRA Consultation on Statutory Fees: Proposals to Recover Costs’ (31 August 2022) <https://assets.publishing.service.gov.uk/media/630f81fdd3bf7f2a2be4b47b/MHRA-Statutory-Fees-Consultation.pdf> accessed 3 February 2024.
[194] Davis and Abraham, Unhealthy Pharmaceutical Regulation (n 180).
[195] MHRA, ‘MHRA Consultation on Statutory Fees’ (n 193).
[196] M Demasi, ‘From FDA to MHRA: Are Drug Regulators for Hire?’ (2022) 377, 01538 BMJ.
[197] Ibid.
[198] Ibid.
[199] Demasi, ‘From FDA to MHRA’ (n 196).
[200] J Lexchin and O O’Donovan, ‘Prohibiting or “Managing” Conflict of Interest? A Review of Policies and Procedures in Three European Drug Regulation Agencies’ (2010) 70, 643 Social Science & Medicine.
[201] MHRA, ‘MHRA Corporate Plan 2023–26’ (n 48).
[202] Lexchin and O’Donovan, ‘Prohibiting or “Managing” Conflict of Interest?’ (n 200).
[203] Davis and Abraham, Unhealthy Pharmaceutical Regulation (n 180).
[204] Hofer and others, ‘Regulatory Policy’ (n 3).
[205] MHRA, ‘Apply for the Early Access to Medicines Scheme’ (n 76).
[206] A Karch and others, ‘Policy Diffusion and the Pro-innovation Bias’ (2019) 69, 83 Political Research Quarterly.
[207] MHRA, ‘MHRA Corporate Plan 2023–26’ (n 48).
[208] MHRA, ‘About Us’ <www.gov.uk/government/organisations/medicines-and-healthcare-products-regulatory-agency/about#our-independent-advisory-bodies> accessed 3 February 2024.
[209] The Human Medicines Regulations 2012 (n 45).
[210] Medicines and Medical Devices Act 2021 (n 42).
[211] S Cruz Rivera and others, ‘Advancing UK Regulatory Science Strategy in the Context of Global Regulation: A Stakeholder Survey’ (2021) 55, 646 Therapeautic Innovation & Regulatory Science.
Image credit: SolStock
Related content

New rules?
Lessons for AI regulation from the governance of other high-tech sectors